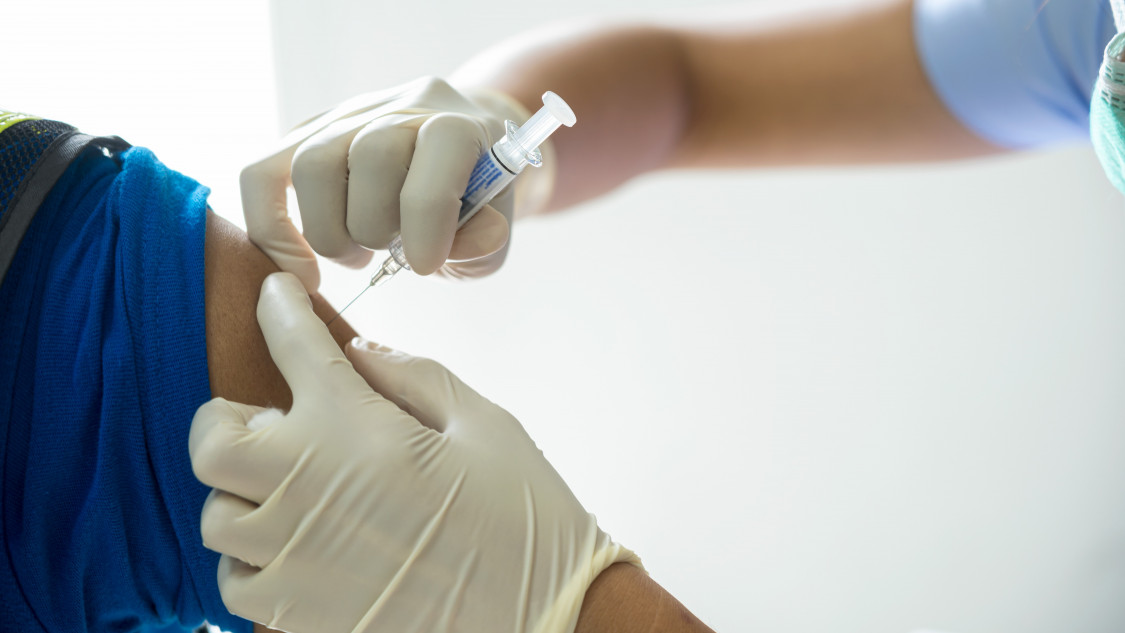
Az Európai Gyógyszerügynökség (EMA) pénteken kiadott, egészségügyi szakembereknek szánt közleményében nem javasolta az AstraZeneca gyógyszergyár koronavírus elleni oltóanyaga második adagjának beadását azoknak, akinek az első adag beadását követően lecsökkent a vérlemezkeszáma, azaz akiknek esetében fennállhat a vérrögképződés esélye.
Az Európai Unió gyógyszerfelügyeleti hatóságának szerepét betöltő ügynökség Humángyógyszer Bizottsága (CHMP) az AstraZeneca svéd-brit gyógyszergyár és az Oxfordi Egyetem közösen kifejlesztett, Vaxzevria nevű vakcinája folyamatos felülvizsgálatának eredményeképpen tett közzé ajánlást, amely szerint elképzelhető az összefüggés az oltóanyag és a nagyon ritka vérrögképződéses esetek között.
Az ajánlás szerint mindazoknál, akiknél fennállhat a vérrögképződés esélye, az AstraZeneca-vakcina első adagjának beadásától számított három héten belül ellenőrizést kell végezni. Noha a vérrögök alacsony vérlemezkeszám miatti kialakulásának esélye nagyon ritka, az EMA azt tanácsolta a szakembereknek, legyenek tisztában az esetleges tünetekkel, és szükség esetén biztosítsanak megfelelő ellátást a pácienseknek.
Az EMA korábban rendkívüli vizsgálatot folytatott a Vaxzevria mellékhatásaival összefüggésben, ugyanis március elején és azóta több ország bejelentette: elővigyázatosságból ideiglenesen felfüggesztik az oltóanyag használatát, miután több olyan esetet is jelentettek, amikor vérrög képződött a beoltottak szervezetében. A korlátozást egyes országok azóta feloldották, mások megerősítették. Az uniós gyógyszerügynökség április elején döntött úgy, hogy a Vaxzevria oltóanyag esetében a vérrögképződés esetleges veszélyét fel kell tüntetni a vakcina „nagyon ritka mellékhatásaként”. Kiemelték: a vakcina előnyei továbbra is felülmúlják a lehetséges mellékhatások kockázatait.
Az Európai Gyógyszerügynökség folyamatosan ellenőrzi az eddig uniós használatra engedélyezett vakcinákat, köztük az AstraZeneca, a Pfizer/BioNtech, a Moderna és a Johnson & Johnson oltóanyagait esetleges mellékhatások felderítésére – közölték.
Az EMA arról is tájékoztatott pénteken, hogy megújította az amerikai Gilead Sciences gyógyszeripari vállalat koronavírus elleni újfajta, remdesivir hatóanyagú, Veklury nevű gyógyszerének feltételes forgalomba hozatali engedélyét. Mint emlékeztettek, a gyógyszert először 2020. június 25-én javasolták jóváhagyásra. Használatát azon 12 éves vagy annál idősebb, tüdőgyulladásban szenvedő betegeknél ajánlották, akiknek kiegészítő oxigén biztosítására van szükségük. A gyógyszerügynökség szakbizottsága azért ajánlotta az engedély meghosszabbítását, mert úgy ítélte meg, hogy a Veklury előnyei továbbra is meghaladják az esetleges kockázatokat – közölték.
Tájékoztattak arról is, hogy az ügynökség Humángyógyszer Bizottsága pénteken befejezte a Sotrovimab nevű, koronavírus-fertőzöttek kezelésére kifejlesztett gyógyszer hatékonyságának vizsgálatát. Az ügynökség arra a következtetésre jutott, hogy a VIR-7831 vagy GSK4182136 kódjelű gyógyszer alkalmazható olyan koronavírus-fertőzött felnőttek és 12 évesnél idősebb serdülők kezelésére, akik nem igényelnek kiegészítő oxigénterápiát, de akiknél fennáll annak a veszélye, hogy a fertőzés miatt súlyos állapotba kerülnek.
A Sotrovimab egy monoklonális antitest, vagyis olyan immunfehérje, melyet úgy terveztek, hogy egy adott struktúrához, jelen esetben a koronavírus-fertőzést okozó vírus, a SARS-CoV-2 tüskefehérjéhez kapcsolódjon. A kapcsolódás létrejöttével a vírusnak a test sejtjeibe való bejutási képessége csökken. A fejlesztők szerint ez várhatóan mérsékli a kórházi kezelés szükségességét a koronavírus-fertőzésben szenvedő betegeknél, illetve az elhalálozásuk kockázatát.
Befejezte az Európai Gyógyszerügynökség (EMA) szakbizottsága a Johnson & Johnson csoportba tartozó Janssen Pharmaceutica-Cilag NV gyógyszeripari vállalat koronavírus elleni vakcinája és a nagyon ritka vérrögképződéses esetek összefüggésében indított vizsgálatát, és kiemelte: hogy ez esetben is az oltóanyag előnyei felülmúlják az esetleges mellékhatások kockázatait – közölte az uniós gyógyszerügynökség pénteken.
Az Európai Unió gyógyszerfelügyeleti hatóságának szerepét betöltő amszterdami ügynökség április első felében kezdte meg a Janssen által az új típusú koronavírus ellen kifejlesztett oltóanyaggal végzett beoltást követően jelentett nagyon ritka vérrögképződéses esetek kivizsgálását, miután az Egyesült Államokban addig négy, később további két erre utaló esetet jelentettek.
Az uniós ügynökség illetékes kockázatértékelő bizottsága (PRAC) összegzésében a vakcina termékinformációjának frissítését javasolta: az oltóanyag esetében a vérrögképződés esetleges veszélyét fel kell tüntetni a vakcina „nagyon ritka mellékhatásaként”.
A termékismertető mostantól tartalmazza azt is, hogy az egészségügyi szakembereknek és az oltásra váróknak tisztában kell lenniük azzal az eshetőséggel, hogy a beoltástól számított három héten belül nagyon ritka esetekben vérrögök képződése, illetve a trombociták számának csökkenése fordulhat elő.
Közölték azt is, hogy az uniós gyógyszerügynökség továbbra is vizsgálja, hogy a Pfizer és a BioNTech gyógyszer- és biotechnológiai cégek Comirnaty névre keresztelt oltóanyaga, valamint a Moderna vállalat koronavírus ellen kifejlesztett vakcinája esetében valószínűsíthető-e a nagyon ritka vérrögképződés előfordulása.
A feltételezett mellékhatásokról szóló jelentésében a szakbizottság úgy vélte, nincs erre utaló jel a sorolt vakcinákkal összefüggésében. A vérrögképződés kialakulásának veszélye rendkívül alacsony, kialakulásának lehetősége alacsonyabb, mint azoknál, akiket nem oltottak be – írták. Az EMA továbbra is szoros figyelemmel kíséri az unió területén ideiglenes használatra engedélyezett valamennyi koronavírus elleni oltóanyaggal kapcsolatos adatot és tapasztalatokat – tették hozzá.
EU officials: a ?? medical team has been granted access to #Saakashvili. No date yet when they will travel to #Georgia. and this is NOT about a transfer of the former President to Poland or elsewhere.
Megadta a vészhelyzeti engedélyt az Egészségügyi Világszervezet (WHO) a kínai Sinopharm oltóanyagának. Az engedély azt jelenti, hogy egy adott oltóanyag hatékony és biztonságos, a szakértői munkacsoport 18 év felettieknek ajánlja, két dózisban.
Az engedély megadását a WHO főigazgatója, Tedrosz Gebrejeszusz sajtótájékoztatón jelentette be. Hozzátette: a világszervezetnek ezzel már hat oltóanyaga van, melyet a COVAX-programban használhat.
Az engedély megadása rendkívül fontos lépés a világ szegényebb országainak. A WHO a koronavírus-járvány alatt indította el az úgynevezett COVAX-kezdeményezést, melynek lényege, hogy egyenlőbb vakcinaelosztást biztosítson, vagyis a szegényebb országok is hozzájuthassanak a koronavírus elleni oltóanyagokhoz. Gebrejeszusz korábban elismerte, hogy a kezdeményezés nem problémamentes, többször vádolta például azzal a gazdagabb országokat, hogy magas áron felvásárolják a vakcinakészleteket. Ezt bizonyítja, hogy áprilisban a világ vakcináinak mindössze 0,3 százaléka került a szegényebb országokba.
A COVAX esetében egy másik lényeges pont, hogy csak olyan vakcinákat oszthatnak ki rajta keresztül az érintett országoknak, melyek már megkapták a WHO engedélyét. Erre azért van szükség, mert míg az Egyesült Államoknak, vagy az Európai Uniónak van megbízható, saját gyógyszerügynöksége, ez más országokra egyáltalán nem igaz.
Az engedélyt eddig öt oltóanyag kapta meg: a Pfizeré, az AstraZenecáé, a CoviShield (az AstraZeneca indiai gyártású változata), a Johnson & Johnsoné és a Modernáé. Mivel azonban ezekre még a nyugati országokban is hatalmas a kereslet, az indiai járványhelyzet miatt pedig a CoviShield vakcinák exportját leállították, nagy szükség van újabb oltóanyagokra. Itt lépett be hatodiknak a kínai gyártású Sinopharm.
A WHO nem hatóság, így engedélye nem kötelező előírás: arról, hogy végül tényleg alkalmazzák-e az adott oltóanyagot, az adott országok egészségügyi hatóságai döntenek.
Ők ugyanúgy megszabhatják azt is, hogy az oltóanyagot csak egy adott korcsoportban alkalmazzák, ez egyébként az Európai Gyógyszerügynökség esetében is hasonlóan működik. Jó példa erre az AstraZeneca oltóanyaga: bár az Európai Gyógyszerügynökség (EMA) 18 év felett biztonságosnak és hatékonynak találta a vakcinát, a vérrögképződéses esetek miatt több uniós tagállam mégis felfüggesztette használatát, vagy csak egy bizonyos korosztályt olt vele.
Megkezdte az Európai Gyógyszerügynökség (EMA) a kínai Sinovac Life Sciences biotechnológiai vállalat által a koronavírus ellen kifejlesztett, inaktivált vírust (Vero Cell) tartalmazó vakcina folyamatos felülvizsgálatát (rolling review) – közölte az Európai Unió gyógyszerfelügyeleti hatóságának szerepét betöltő, amszterdami székhelyű uniós ügynökség kedden.
Az uniós ügynökség közleménye szerint döntése a szakvizsgálat megkezdéséről a laboratóriumi kutatások és a klinikai vizsgálatok előzetes, kedvezőnek mutatkozó eredményein alapul. A tanulmányok azt sugallják, hogy az inaktivált vírust tartalmazó vakcina olyan antitestek termelését váltja ki, amelyek a SARS-CoV-2-t, azaz a Covid-19-et okozó vírust támadják meg, és elősegíthetik a betegség elleni védekezést.
Az EMA Humángyógyszer Bizottsága (CHMP) eljárása során értékelni fogja a vakcina hatékonyságára és biztonságosságára, valamint minőségére vonatkozó adatokat, hogy megbizonyosodjanak róla, hogy az oltóanyag előnyei felülmúlják-e az esetleges kockázatokat. A folyamatos felülvizsgálat addig folytatódik, amíg elegendő bizonyíték nem áll rendelkezésre a forgalomba hozatali engedély hivatalos kérelmének beadásához.
Az EMA közölni fogja a vakcináról kialakított véleményét, illetve azt, ha a gyártó benyújtja az oltóanyag forgalomba hozatali engedély iránti kérelmét – közölték.
Az EMA tájékoztatása szerint az oltóanyag elősegíti, hogy a védőoltás után az emberi szervezet immunrendszere az inaktivált vírust idegen anyagként azonosítsa, és természetes védekezésként antitesteket termeljen ellene. Ha az oltott személy később kapcsolatba kerül a koronavírussal, immunrendszere felismeri a vírust, és készen áll arra, hogy védelmet nyújtson a koronavírus ellen. Az inaktivált vírus nem okozhat betegséget – tették hozzá.
Az Európai Gyógyszerügynökség (EMA) kedden közölte, hogy csak rendkívül ritka esetekben állhat fenn kapcsolat a Johnson & Johnson csoportba tartozó Janssen Pharmaceutica-Cilag NV gyógyszeripari vállalat koronavírus elleni vakcinája és az ugyancsak nagyon ritka vérrögképződéses esetek között. Az eddigi eredmények azonban azt mutatják, hogy az oltóanyag előnyei felülmúlják a lehetséges mellékhatások kockázatait.
Az Európai Unió gyógyszerfelügyeleti hatóságának szerepét betöltő amszterdami ügynökség április első felében kezdte meg a Janssen által az új típusú koronavírus ellen kifejlesztett oltóanyaggal végzett beoltást követően jelentett nagyon ritka vérrögképződéses esetek kivizsgálását, miután az Egyesült Államokban addig négy, később további két erre utaló esetet jelentettek.
Az uniós ügynökség illetékes kockázatértékelő bizottsága (PRAC) véleménye szerint ugyanakkor szükséges a vakcina termékinformációjának frissítése. Javaslatuk szerint az oltóanyag esetében a vérrögképződés esetleges veszélyét fel kell tüntetni a vakcina „nagyon ritka mellékhatásaként”.
„Az egészségügyi szakembereknek és az oltásra váróknak tisztában kell lenniük azzal az eshetőséggel, hogy a beoltástól számított három héten belül nagyon ritka esetekben vérrögök képződése, illetve a trombociták számának csökkenése fordulhat elő” – fogalmaztak.
A jelentett esetek többsége 60 év alattiaknál, többségükben nők között fordult elő az oltást követő két héten belül. A jelenleg rendelkezésre álló bizonyítékok alapján azonban konkrét kockázati tényezőt nem azonosítottak – emelték ki.
Megjegyezték, hogy a vizsgált esetek hasonlóságot mutatnak az AstraZeneca gyógyszergyár és az Oxfordi Egyetem Vaxzevria névre keresztelt oltóanyagánál jelentkező nagyon ritka rendellenességhez. A vérrögképződés szokatlan módon az agy sinus vénáiban, a hasi vénákban és az artériákban fordult elő. A vérrögképződés egyik elfogadható magyarázata, hogy a vakcina immunválaszt vált ki, amely hasonló állapothoz vezet, mint amely a heparinnal kezelt betegeknél néha előfordul. Az úgynevezett heparin indukálta trombocitopénia (HIT) a heparin kezelés egyik legsúlyosabb szövődménye, mely adott esetben tromboemboliás komplikációkhoz vezet.
Hozzátették, az ügynökség illetékes kockázatértékelő bizottsága a vérrögképződés esetleges veszélye esetén az azonnali szakorvosi konzultáció fontosságát hangsúlyozza. Vérrögképződésre, vagy a vérlemezkék alacsony számára utaló jelek korai felismerése elkerülheti ugyanis az esetleges szövődmények kialakulását. A beoltottaknak haladéktalanul orvoshoz kell fordulniuk, ha esetükben légszomj, mellkasi, vagy hasi fájdalom, fejfájás, vagy homályos látás tünetei jelentkeznek, illetve akkor, ha duzzanatot figyelnek meg a lábban, vagy apró vérfoltok jelennek meg a bőr alatt az injekció beadásának helyén kívül.
Végezetül arra emlékezettek, hogy az amerikai egészségügyi hatóságok a beoltás után néhány nappal hat esetben jelentkező vérrögképződéses rendellenességek nyomán a Johnson & Johnson koronavírus elleni, egydózisú vakcinája alkalmazásának felfüggesztését javasolták. Az esetek közül egy halálos kimenetelű volt. Az Egyesült Államokban mostanáig több mint 7 millió adag Johnson & Johnson-vakcinát adtak be.
Az EMA az összes többi Európai Unióban engedélyezett oltóanyaghoz hasonlóan továbbra is figyelemmel kíséri a Jansen vakcinájának biztonságosságát és hatékonyságát, a legfrissebb információkat közli a nyilvánossággal – tették hozzá.
(MTI)
[type] => post
[excerpt] => Az Európai Gyógyszerügynökség (EMA) kedden közölte, hogy csak rendkívül ritka esetekben állhat fenn kapcsolat a Johnson & Johnson csoportba tartozó Janssen Pharmaceutica-Cilag NV gyógyszeripari vállalat koronavírus elleni vakcinája és az ugyancsak...
[autID] => 5
[date] => Array
(
[created] => 1618994880
[modified] => 1618956704
)
[title] => Az Európai Gyógyszerügynökség bejelentette, lehetséges a kapcsolat a Johnson Johnson vakcinája és a vérrögképződéses esetek között
[url] => https://life.karpat.in.ua/?p=53424&lang=hu
[status] => publish
[translations] => Array
(
[hu] => 53424
[uk] => 53411
)
[aut] => gygabriella
[lang] => hu
[image_id] => 44300
[image] => Array
(
[id] => 44300
[original] => https://life.karpat.in.ua/wp-content/uploads/2021/01/jonson9.jpg
[original_lng] => 141334
[original_w] => 1000
[original_h] => 563
[sizes] => Array
(
[thumbnail] => Array
(
[url] => https://life.karpat.in.ua/wp-content/uploads/2021/01/jonson9-150x150.jpg
[width] => 150
[height] => 150
)
[medium] => Array
(
[url] => https://life.karpat.in.ua/wp-content/uploads/2021/01/jonson9-300x169.jpg
[width] => 300
[height] => 169
)
[medium_large] => Array
(
[url] => https://life.karpat.in.ua/wp-content/uploads/2021/01/jonson9-768x432.jpg
[width] => 768
[height] => 432
)
[large] => Array
(
[url] => https://life.karpat.in.ua/wp-content/uploads/2021/01/jonson9.jpg
[width] => 1000
[height] => 563
)
[1536x1536] => Array
(
[url] => https://life.karpat.in.ua/wp-content/uploads/2021/01/jonson9.jpg
[width] => 1000
[height] => 563
)
[2048x2048] => Array
(
[url] => https://life.karpat.in.ua/wp-content/uploads/2021/01/jonson9.jpg
[width] => 1000
[height] => 563
)
[full] => Array
(
[url] => https://life.karpat.in.ua/wp-content/uploads/2021/01/jonson9.jpg
[width] => 1000
[height] => 563
)
)
)
[video] =>
[comments_count] => 0
[domain] => Array
(
[hid] => life
[color] => red
[title] => Життя
)
[_edit_lock] => 1618949242:5
[_thumbnail_id] => 44300
[_edit_last] => 5
[views_count] => 6926
[_hipstart_feed_include] => 1
[_oembed_f7e2f8ef52e510b70fa48ed401760f39] =>
[_oembed_time_f7e2f8ef52e510b70fa48ed401760f39] => 1627024251
[_algolia_sync] => 508054283000
[labels] => Array
(
)
[categories] => Array
(
[0] => 41
[1] => 4383
[2] => 49
[3] => 39
)
[categories_name] => Array
(
[0] => Cikkek
[1] => COVID-19 HU
[2] => Hírek
[3] => Világ
)
[tags] => Array
(
[0] => 43509
[1] => 36440
[2] => 30447
)
[tags_name] => Array
(
[0] => Európai Gyógyszerügynökség (EMA)
[1] => Johnson & Johnson
[2] => mellékhatás
)
)
[5] => Array
(
[id] => 52989
[content] =>
Az AstraZeneca koronavírus elleni vakcinájának egy második lehetséges mellékhatását vizsgálja az Európai Gyógyszerügynökség (EMA) kockázatelemző bizottsága (PRAC).
A PRAC egy korábbi vizsgálata megállapította, hogy a brit–svéd gyógyszergyártó egy ideje új néven forgalmazott koronavírus elleni vakcinájának egy nagyon ritka mellékhatása, hogy az alkalmazása utáni két hétben vérrögképződéssel, ezzel párhuzamosan pedig a vérlemezkék (trombociták) számának kóros csökkenésével járhat.
A bizottság most egy másik, az AstraZeneca oltásával kapcsolatos probléma kapcsán nyomoz.
Eddig öt esetben számoltak be arról, hogy a vakcinával beoltottaknál kapilláris szivárgás szindróma lépett fel. A PRAC arra kíváncsi, van-e összefüggés az oltóanyag, és a megbetegedések között – írja a Brussels Times.
A PRAC munkáját nem könnyíti meg, hogy magáról a kapilláris szivárgás szindrómáról sem tudunk sokat. A betegséget – amelynek során a vérplazma a hajszálerekből a környező szövetekbe szivárog, kórosan alacsony vérnyomást, és a vér besűrűsödését, valamint ödémákat okozva – először 1960-ban írták le. Ha nem kezelik, a szervek leállásához, és halálhoz is vezethet. A kapilláris szivárgás szindróma azonosítását nem könnyíti meg, hogy tünetei hasonlítanak a szepsziséhez (vérmérgezés), ezért előfordulhat, hogy korábbi esetek elkerülték a szakemberek figyelmét – jegyzi meg a The Conversationben publikált írásában Peter Abel, a University of Central Lanchasire tanára.
Az EMA kockázatelemzői a Johnson&Johnson vakcinájával kapcsolatos jelentéseket is vizsgálnak. Az uniós használatra még márciusban engedélyezett, de Európában eddig még tömegesen nem használt vakcinát kapottaknál négy esetben alakult ki vérrög-képződés, de az oltás és a kóros állapot kialakulása közötti összefüggés nem bizonyított. Az oltóanyag használatát az Egyesült Államokban és a Dél-afrikai köztársaságban felfüggesztették.
Az Európai Gyógyszerügynökség (EMA) vizsgálja, hogy van-e összefüggés a Johnson & Johnson vakcinájának használata és a vérrögképződés között. Négy páciensnél keletkeztek vérrögök a Janssen oltás beadása után, egyikük meg is halt.
Az EMA közleményében azt írta, hogy négy olyan esetet is találtak, ahol a Janssen nevű oltóanyag beadása után alacsony vérlemezkeszámú vérrögök keletkeztek, egy páciens bele is halt a következményekbe.
Az Európai Gyógyszerügynökség március 11-én engedélyezte a Johnson & Johnson egydózisú oltóanyagát, amely 67 százalékos hatékonysággal előzi meg a fertőzést.
Magyarország összesen 4,36 millió adagot kötött le a vakcinából. Müller Cecília országos tiszti főorvos az operatív törzs március 31-ei sajtótájékoztatóján azt mondta, hogy április második felében érkezhetnek a Janssen vakcinák Magyarországra.
Az Európai Gyógyszerügynökség az AstraZeneca vakcináját is vizsgálja a trombózisos esetek miatt. Szerdán azt közölték, hogy lehet kapcsolat a vérrögképződés és az oltóanyag beadása között, de álláspontjuk szerint továbbra is megéri oltani vele. A WHO szerint a brit–svéd gyártó védőoltása és a vérrögképződés ritka formája közötti kapcsolat lehetséges, de nem megerősített.
Összefüggés lehet az AstraZeneca-oltás és a nagyon kisszámú, véralvadással kapcsolatos esetek között – jelentette be az Európai Gyógyszerügynökség (EMA) szerdán a gyógyszerkockázat-értékelő bizottságának (PRAC) újabb vizsgálata után.
Ennek ellenére az EMA továbbra is ajánlja az AstraZeneca-oltások használatát, mivel annak előnyei jóval meghaladják a hátrányait.
Az Európai Gyógyszerügynökség szerint ugyanakkor az alacsony trombocitaszámot az AstraZeneca nagyon ritka mellékhatásaként kell feltüntetni, fel kell hívni az egészségügyi dolgozók és a beoltottak figyelmét arra, hogy az oltást követő két hétben vérrög képződhet a szervezetükben.
A vérrögképződéssel járó megbetegedés, amely a trombociták számának alacsony számával áll összefüggésben néhány esetben, eddig igen alacsony arányban fordult elő az AstraZeneca cég termékével beoltottak körében. Az EMA tájékoztatása szerint az Európai Gazdasági Térség országaiban és Nagy-Britanniában több mint 25 millió embert oltottak be AstraZeneca-vakcinával, és mindössze 62 esetben számoltak be agyi, 24 további alkalommal pedig a belső szerveket érintő trombózisról, és közülük 18-an vesztették életüket.
Emer Cooke, az EMA igazgatója az eredményeket ismertető sajtótájékoztatóján hangsúlyozta, hogy ha valakinek AstraZeneca-oltást ajánlanak, azt el kell fogadni, ugyanis sokkal nagyobb esélye van annak, hogy valaki a koronavírusban meghal, mint annak, hogy a vakcina mellékhatásaként életét veszti.
Noha Cooke elismerte, hogy az eddig jelentett trombózisos esetek többségében 60 év alatti nők voltak az érintettek, azt is kifejtette, hogy a rendelkezésre álló adatok alapján végzett vizsgálat nem igazolta, hogy bizonyos életkorú vagy nemű betegek hajlamosabbak lennének a vérrögképződésre, ahogy korábbi betegségek sem jelezhetik a kockázatot előre. Annyit tudtak csak megállapítani, hogy az ismert esetekben az AstraZeneca-oltásra a betegek olyan immunválaszt adtak, mint amilyet ritka esetekben a heparin véralvadásgátló vált ki.
A kockázat összehasonlításaként dr. Peter Arlett, az EMA adatelemzői munkacsoportjának vezetője elmagyarázta: 10 ezer fogamzásgátló szedő nőből évente 4-nél alakul ki trombózis.
Marco Cavaleri, az EMA oltásokat vizsgáló munkapcsoportjának vezetője egy olasz lapnak adott interjúban kedden úgy fogalmazott: „a véleményem szerint már kimondhatjuk, hogy a nagyon ritka trombózisos esetek kapcsolatban vannak az oltással”, és azt is hozzátette: tény, hogy főleg fiatal beoltottak között magasabb az agyi trombózis esetszáma, mint várnák.
Márciusban az AstraZeneca-oltás alkalmazását több ország felfüggesztette, miután felvetődött, hogy az oltóanyag vérrögképződést okozhat. Ezt követően az EMA és az Egészségügyi Világszervezet (WHO) is megvizsgálta a vakcinát, és arra jutott, hogy az oltóanyag hatékony és biztonságos.
Az uniós gyógyszerügynökség vizsgálatot folytat a vérrögképződéses esetek, illetve az ez idáig uniós használatra engedélyezett valamennyi vakcina, köztük a Pfizer/BioNTech, a Moderna és a Johnson & Johnson oltóanyagai esetleges mellékhatásainak kiderítésére is.
A vizsgálatok továbbra sem azonosítottak kockázati tényezőket sem életkorral, sem pedig a vérrögképződéssel összefüggő rendellenességek korábban jelzett nagyon ritka eseteivel kapcsolatban az AstraZeneca gyógyszergyár koronavírus elleni vakcinája esetében – közölte szerdán az Európai Gyógyszerügynökség (EMA), írja a Portfolio.
Az Európai Unió gyógyszerfelügyeleti hatóságának szerepét betöltő amszterdami székhelyű ügynökség közleményében kiemelte: a brit-svéd gyógyszergyártó és az Oxfordi Egyetem oltóanyaga, valamint a nagyon kisszámú, véralvadással kapcsolatos esetek közötti összefüggés továbbra sem bizonyított. Mivel azonban lehetséges, a vakcina esetleges mellékhatásainak elemzése folytatódik.
Kiemelték: a vakcina előnyei továbbra is felülmúlják a lehetséges mellékhatások kockázatait. Az ügynökség illetékes kockázatértékelő bizottsága (PRAC) jövő hétre tervezett havi ülése után naprakész információkat tesz közzé az AstraZeneca oltóanyagának vizsgálatának állásáról – tették hozzá.
A vérrögképződéssel járó megbetegedés, amely a trombociták számának veszélyes mértékű emelkedését is előidézte néhány esetben, eddig igen alacsony arányban fordult elő az AstraZeneca cég termékével beoltottak körében. Az EMA korábbi tájékoztatása szerint az Európai Unió országaiban és Nagy-Britanniában több mint 17 millió, AstraZeneca-vakcinával beoltott között pár tucat vérrögképződéses esetről számoltak be.
Az uniós gyógyszerügynökség vizsgálatot folytat a vérrögképződéses esetek, illetve az ez idáig uniós használatra engedélyezett valamennyi vakcina, köztük a Pfizer/BioNtech, a Moderna és a Johnson & Johnson oltóanyagai esetleges mellékhatásainak kiderítésére.
Az EMA azért végez rendkívüli vizsgálatot, mert március elején és azóta több ország bejelentette: elővigyázatosságból ideiglenesen felfüggesztik az AstraZeneca oltóanyagának használatát, miután több olyan esetet is jelentettek, amikor vérrög képződött a beoltottak szervezetében. A korlátozást egyes országok azóta feloldották, mások megerősítették.